
EL POLVO DEL SAHARA

Historia breve de «LA LIMPIEZA»
junio 2, 2020
LOS 8 PRINCIPIOS DE UNA CASA SALUDABLE.
junio 28, 2020

El Desierto del Sahara
Los aerosoles son partículas suspendidas líquidas, sólidas o de fases múltiples de materia condensada en un medio gaseoso. Varían en su composición (es decir, biológica o mineral), tamaño (0,001-100 μm) y forma (Kulkarni et al., 2011). En los últimos años, los aerosoles han recibido cada vez más atención como importantes agentes del cambio climático (Solomon et al., 2011).
Las regiones áridas como los desiertos son la principal fuente de partículas de polvo mineral en la atmósfera. Allí, las partículas de polvo son levantadas por la actividad de las tormentas y transportadas en forma de aerosoles a altitudes superiores a los 5.000 metros sobre el nivel del mar (m.s.n.m.) (Prospero et al., 2005). El desierto del Sahara en África es la principal fuente de aerosoles minerales del mundo (Goudie y Middleton, 2006) y se ha estimado que contribuye con 630-710 Mt a la atmósfera cada año (D’Almeida, 1986). Aproximadamente el 60% de las emisiones de polvo sahariano se transportan hacia el sur, el 25% hacia el oeste hasta el Atlántico, el 5% hacia el este hasta el Oriente Medio y el 10% hacia el norte hasta Europa (Shao et al., 2011).
Hasta el 25% de la masa total mundial de aerosoles está compuesta por partículas biológicas, o bioaerosoles (Jaenicke, 2005). Las primeras descripciones de los bioaerosoles fueron proporcionadas por Charles Darwin, quien observó «67 formas orgánicas diferentes» en las partículas de polvo fino que se depositaron en el Beagle durante su viaje a través del Océano Atlántico en 1833 (Darwin, 1846). En general, los bioaerosoles contienen fragmentos de piel, fibras de pelo, cristales de proteínas, polen, fragmentos de plantas, esporas, virus, algas, hongos y bacterias, y pueden flotar libremente o adherirse a aerosoles minerales (Després et al., 2012; Deleon-Rodriguez et al., 2013). Los bioaerosoles como virus, bacterias y hongos han cobrado cada vez más interés por su potencial para propagar patógenos a grandes distancias (Prospero et al., 2005; Griffin, 2007; Polymenakou, 2012). También se han relacionado con los procesos atmosféricos (Morris et al., 2011; Þantl-Temkiv et al., 2013). Por ejemplo, las bacterias transportadas por el aire pueden servir como núcleos de hielo durante las precipitaciones de nieve (Amato et al., 2007b; Bowers et al., 2009; Hiranuma et al., 2015).
El transporte a larga distancia de bacterias viables en conjunción con los aerosoles minerales puede representar una forma de que las bacterias colonicen nuevos entornos y contribuyan a aumentar la diversidad en hábitats terrestres y acuáticos remotos (Barton et al., 2010). De hecho, se demostró que las bacterias transportadas por el aire de las partículas de polvo del Sahara (SD) recogidas en el Beagle de Darwin en el siglo XIX eran viables 150 años más tarde (Gorbushina et al., 2007). Sin embargo, todavía no se sabe con certeza en qué medida los organismos siguen siendo viables o activos después de la dispersión aérea, así como su potencial para establecer una colonia después de la deposición (Pointing and Belnap, 2012). Debido a las duras condiciones asociadas al transporte aéreo (por ejemplo, estrés de desecación, exposición a los rayos ultravioleta, condiciones oligotróficas y bajas temperaturas) (Smith et al., 2009), sólo los taxones especialmente adaptados son capaces de sobrevivir el viaje a un nuevo entorno. La esporulación o la pigmentación podrían ayudar a proteger a las bacterias contra las duras condiciones que prevalecen durante el transporte (Tong y Lighthart, 1997). Por ejemplo, la Janthinobacterium es una bacteria que se detecta comúnmente en las muestras de aire y produce un pigmento indigo-púrpura (Fahlgren et al., 2010).
Hasta la fecha, se han tomado muestras de bacterias transportadas por el aire del desierto del Sáhara a lo largo de la costa de Grecia y el sudeste del Mediterráneo (Griffin et al., 2007; Polymenakou et al., 2008; Katra et al., 2014), en España (Sánchez De La Campa et al., 2013; Barberán et al., 2014) y en el Caribe (Griffin et al., 2003; Prospero et al., 2005). El muestreo de bacterias en el aire puede realizarse mediante técnicas como la filtración, la impactación, la succión, el pinzamiento y la precipitación electrostática, cada una de las cuales presenta diferentes ventajas y desventajas para los análisis biológicos posteriores específicos (por ejemplo, las técnicas basadas en la PCR) (Mandal y Brandl, 2011). La recogida de aerosoles, ya sea en el interior o en el exterior, requiere la comprensión de los principios físicos que influyen en la recogida de partículas en suspensión que pueden causar sesgos cuantitativos y cualitativos (Després et al., 2012). En particular, la recolección de bioaerosoles requiere procedimientos especiales de manipulación para evitar que se dañen las células durante el proceso de recolección (por ejemplo, el estrés por deshidratación) (Després et al., 2012). Por último, el momento y la duración de los episodios de polvo sahariano siguen siendo difíciles de predecir, lo que dificulta la capacidad de los investigadores para recuperar un volumen de aire adecuado para los análisis microbiológicos.
Las bacterias depositadas en la nieve permiten a los investigadores eludir los inconvenientes del muestreo del aire. Se preservan tanto la integridad física como la viabilidad potencial de las células bacterianas, ya que se recogen a temperaturas de congelación desde el interior de la capa de nieve. Las ubicaciones a gran altitud por encima de la influencia de la capa límite planetaria son ideales para detectar las partículas de polvo del Sáhara (SD) depositadas porque la contaminación potencial de los aerosoles de origen antropogénico después de la deposición es mínima
La mayoría de los estudios de SDE realizados en los Alpes se han centrado en los aspectos mineralógicos y químicos (Sodemann y otros, 2006; Thevenon y otros, 2009); todavía faltan análisis microbiológicos. Los primeros intentos en este sentido fueron realizados por González-Toril y otros (2009) y Chuvochina y otros (2011a) en el glaciar del Mont Blanc. Sin embargo, en estos estudios no se presentaron detalles sobre los parámetros físico-químicos de la nieve, la posición de las capas de polvo del Sáhara (capas SD) dentro del manto de nieve o las características geoquímicas de las partículas de polvo. A pesar de la valiosa contribución de estos estudios preliminares, la baja profundidad de secuenciación alcanzada por las pequeñas bibliotecas de clones dificulta la obtención de conclusiones sólidas sobre las estructuras de las comunidades bacterianas asociadas a las SDE. Además, los conocimientos son todavía limitados en cuanto a la viabilidad y la actividad metabólica de las bacterias transportadas desde el desierto del Sáhara a los Alpes.
La región de Jungfraujoch, situada en los Alpes suizos a unos 3600 m de altitud, se encuentra por encima de la influencia de la capa límite planetaria y, por lo tanto, es un lugar muy ventajoso para estudiar los aerosoles transportados desde el desierto del Sáhara a los Alpes europeos. Los SDE son monitoreados regularmente en tiempo real en la estación meteorológica de Jungfraujoch. Estos eventos son muy comunes en primavera (marzo-junio) y en otoño (octubre-noviembre) pero raros en verano e invierno (Collaud Coen y otros, 2004; Papayannis y otros, 2008; Conen y otros, 2015; Flentje y otros, 2015). Sin embargo, una SDE invernal excepcionalmente duradera (40 h) depositó partículas de polvo procedentes del desierto del Sáhara en la nieve de los Alpes suizos en febrero de 2014. Las partículas de SDE y las bacterias asociadas fueron cubiertas por nieve fresca 3 días después de la deposición. Esta rara SDE invernal ofreció la oportunidad única de tomar muestras de una capa de SD bien conservada dentro del manto de nieve. Además de la SDE de invierno, una SDE de primavera más corta (22 h) en mayo de 2014 depositó partículas de polvo justo antes del deshielo. Juntos, estos depósitos permiten la comparación de las SDE que se producen en diferentes estaciones y que se originan en diferentes regiones del desierto del Sahara.
El objetivo de este estudio fue caracterizar las comunidades bacterianas dentro de las capas de SD preservadas en la nieve mediante una combinación de secuencias e incubaciones de alto rendimiento para la actividad metabólica microbiológica utilizando MiSeq Illumina® y Biolog EcoPlate™, respectivamente. Comparamos diferentes capas de un perfil de nieve tomado en Jungfraujoch en el que pudimos distinguir las capas de nieve que contienen partículas de SD de las capas de nieve limpia adyacentes (capas CS). Presentamos el primer estudio para lograr una profundidad de secuenciación y cobertura de alta resolución para las bacterias asociadas a la SD en combinación con amplios análisis in situ y de laboratorio de las características fisicoquímicas y geoquímicas de la nieve y de las partículas que la componen.
Ir a
Materiales y métodos
Descripción del lugar y recogida de muestras
El muestreo se realizó en la región de Jungfraujoch (46°33′11″N/8°0′17″E, 3621 m s.n.m.) en un campo de nieve entre la Estación Meteorológica Mundial de Jungfraujoch de alta montaña (No. 06730) y el refugio alpino Mönchsjochhütte.
Una zanja vertical fue excavada por una máquina pisanieves a una profundidad de 220 cm. por debajo de la superficie de la nieve el 8 de junio de 2014 a las 08:00 CEST. Se seleccionaron tres perfiles de nieve (A, B y C) a 100 cm de distancia entre sí para el muestreo en el lado sombreado de la zanja. De los perfiles de nieve se tomaron tres réplicas para análisis fisicoquímicos y microbiológicos, cortando una zanja fresca en la nieve con un cuchillo esterilizado con un 70% de etanol. En cada perfil se cortó un bloque de nieve de 3,5 × 13 × 13 cm a siete profundidades (J0: -25 cm, J1: -80 cm, J2: -120 cm, J3: -145 cm, J4: -150 cm, J5: -155 cm, J6: -170 cm, y J7: -190 cm) y almacenados en una bolsa de congelador estéril (Figura suplementaria 1C).
Las muestras se almacenaron en una caja de hielo en el campo y se transportaron al laboratorio, donde se almacenaron a 0°C. La nieve se derritió lentamente y posteriormente se trasladó a botellas de vidrio Schott de laboratorio esterilizadas en autoclave y almacenadas a 0°C hasta su posterior análisis (volumen total entre 300 y 480 ml por muestra de campo).
Parámetros físico-químicos
Los parámetros físicos como la temperatura y la densidad de la nieve se midieron in situ a lo largo del perfil de la nieve a intervalos de 10 y 20 cm, respectivamente. La temperatura se midió con un sensor de temperatura Testo 925 (Testo AG, Mönchaltdorf, Suiza). La densidad se midió con un método de pesaje de la nieve (Goodison et al., 1981). La conductividad y el pH se midieron en agua de fusión homogéneamente mezclada a temperatura ambiente con un medidor universal de bolsillo Multi 350i (WTW, Weilheim, Alemania) siguiendo las instrucciones del fabricante.
Cálculo de la trayectoria hacia atrás
Las trayectorias hacia atrás de las masas de aire se calcularon utilizando el Modelo de Velocidad Vertical NOAA HYSPLIT y los datos meteorológicos GDAS1 (Draxler y Rolph, 2003). Los ESD fueron detectados por el Instituto Paul-Scherrer (PSI) utilizando el método de la ETI del nefelómetro aSSA BR en el Jungfraujoch como se describe en Collaud Coen et a( 2004). Evaluamos la intensidad de las ESD considerando el área de la dispersión de albedo negativa.
Datos meteorológicos
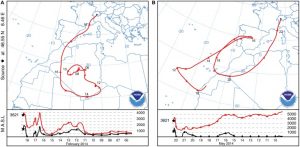
La temperatura máxima diaria y mínima nocturna del aire a 2 m sobre el suelo fue medida en Jungfraujoch (46°32′51″N/7°59′07″E; 3580 m s.n.m.). No hay datos de altura de la nieve disponibles en Jungfraujoch. Sin embargo, la altura de la nieve se evaluó promediando los datos de dos estaciones meteorológicas a 8,6 km al noroeste y 15 km al sudeste de Jungfraujoc
h, respectivam
ente: Männlichen (46°36′47″N/7°56′27″E; 2343 m s.n.m.) y Eggishorn (46°25′36″N/8°05′34″E; 2927 m s.n.m.). Los datos fueron suministrados por la Oficina Federal de Meteorología y Climatología MeteoSwiss (IDAweb).
Caracterización y cuantificación de las partículas de polvo
El agua de fusión (5 ml) que contenía partículas de polvo homogén
eamente suspendidas se filtró a través de un filtro de policarbonato GTTP de 0,2 μm con una bomba de vacío (Millipore, Billerica, MA, USA). Las muestras que contenían altas concentraciones de polvo (J0 y J4) fueron diluidas 1:10 para reducir la densidad de las partículas en el filtro. El mismo procedimiento se aplicó a tres muestras de lecho de roca muestreadas a menos de 200 m de los perfiles de nieve (Rock1: 46°33′9″N/8°0′9″E; Rock2: 46°33′16″N/8°0′27″E, Rock3: 46°33′16″N/8°0′20″E) después de haber sido molida a polvo.
Las partículas de polvo fueron caracterizadas por microscopía electrónica de barrido (SEM) (Jeol 6390LA) en el Instituto de Geoquímica y Petrología (ETH, Zurich). Los análisis químicos de las partículas de las muestras recubiertas de carbono montadas en filtros de policarbonato se obtuvieron mediante espectroscopia de rayos X de energía dispersiva (SEM-EDX), utilizando un detector EDS de estado sólido combinado con el software científico Thermo NSS3 en condiciones de alto vacío. El SEM fue equipado inicialmente con un filamento W que fue posteriormente cambiado a un cristal LAB6. Para todos los análisis se empleó una tensión de aceleración de 15 kV con una corriente del haz de 2,5 nA y un diámetro de sonda de 1 μm. La composición química de cada partícula se midió durante 15 s de tiempo vivo. Debido al pequeño tamaño de las partículas de polvo, el volumen de excitación excedía ocasionalmente el tamaño de las partículas de polvo, lo que dio lugar a artefactos analíticos que no pudieron ser eludidos. Por lo tanto, todos los análisis se normalizan a una base libre de carbono.
Las fases minerales se definieron sobre la base de un gráfico modificado de Moreno (2003): cuarzo y silicatos de bajo contenido en Al (x: 1,0-1,3, y: 0,0-0,2); feldespatos (x: 0. 8-1,2, y: 0,2-0,5); illita (x: 1,3-1,6, y: 0,6-0,9); caolinita (x: 1,6-2,5, y: 0,85-1,2); montmorillonita (x: 1,3-1,5, y: 0,4-0,6).
Las partículas orgánicas e inorgánicas se cuantificaron utilizando un citómetro de flujo BD Accuri C6 (BD Biosciences, San José, EE.UU.). El agua de fusión (200 μl) se fijó con glutaraldehído (Sigma-Aldrich, Buchs, Suiza), se tiñó con 2 μl de Sybr® Green (Life Technologies, Zug, Suiza) y se incubó durante 10 min a 37°C.
Aislamiento del ADN total
En un intento por mejorar la recuperación del ADN de las bacterias formadoras de esporas y compensar las bajas concentraciones de biomasa, se aplicaron dos métodos de extracción de ADN ligeramente diferentes.
Método MOBIO UltraClean: 100 ml de agua de fusión mezclada homogéneamente se filtraron a través de una membrana de nylon estéril de 0,2 μm con un sistema de filtración al vacío (modelo 87006-076, VWR, PA, EE.UU.) (Figura suplementaria 1D). El ADN total se aisló utilizando el kit de aislamiento de ADN MOBIO UltraClean™ (Mobio Laboratories, CA, EE.UU.) de acuerdo con las instrucciones del fabricante con las siguientes modificaciones: los filtros se lavaron durante la noche en un tubo (Falcon 15) con 60 μl de solución S1, 600 μl de solución IRS y 400 μl de solución de perlas. Para separar completamente las partículas de los filtros, los tubos se centrifugaron horizontalmente durante 30 minutos a máxima velocidad. A continuación, los tubos fueron sonicados durante 10 s en un baño de agua a temperatura ambiente e inmediatamente se colocaron en hielo durante 20 s. La sonicación se repitió dos veces. Finalmente, el líquido se transfirió a los tubos que contenían perlas, se incubó durante 10 min a 65°C y se procesó siguiendo las instrucciones del fabricante.
Método MOBIO PowerWater: El volumen restante (variable en cada muestra entre 20 y 280 ml) de agua de fusión mezclada homogéneamente se filtró a través de una membrana de nylon estéril de 0,2 μm con un sistema de filtración al vacío (modelo 87006-076, VWR). Debido a la obstrucción, se utilizaron dos filtros para las muestras que contenían partículas SD. Los filtros fueron cortados en dos m
itades. Una mitad de cada filtro se utilizó para la extracción utilizando el MOBIO PowerWater™ Kit de Aislamiento de ADN (Laboratorios Mobio) de acuerdo con las instrucciones del fabricante con las siguientes modificaciones: Los tubos de microesferas PowerWater® se calentaron a 65°C durante 10 min, como indica el método de lisis alternativo. A continuación, los tubos fueron agitados horizontalmente durante 1 h a máxima velocidad. El ADN fue eluido en 50 μl PW6 y almacenado a -20°C.
El ADN de las tres extracciones (1 extracción con el método MOBIO UltraClean y 2 extracciones con el método MOBIO PowerWater) se unió antes de continuar el procesamiento.
El ADN extraído se cuantificó con el Fluorómetro Qubit® dsDNA HS Assay Kit y se normalizó al volumen total del agua de fusión filtrada.
Amplificador de ciclo bajo PCR
La secuenciación de MiSeq Illumina® se realizó con amplificación de ciclo bajo, indexación Nextera según las instrucciones del Centro de Diversidad Genética (GDC; ETH Zurich, Suiza).
Para la amplificación por PCR, se utilizó el cebador universal hacia adelante Bakt_341F (Número de adhesión pB-03844) 5′-CCTACGGNGGCWGCAG-3′ y el cebador universal invertido Bakt_805R (pB-03845): 5′-GACTACHVGGGTATCTAATCC-3′ (Herlemann et al., 2011) se modificaron para la posterior secuenciación MiSeq Illumina® de la región hipervariable V3-V4 del gen del ARNr 16S añadiendo el saliente (cursiva) y una inserción de cero a tres nucleótidos entre el saliente y la secuencia del iniciador .
Cada muestra replicada se amplificó en cuatro reacciones de PCR separadas con los cuatro pares de cebadores diferentes (fs0, fs1, fs2 y fs3). 20 μl de mezcla de reacción de PCR se compuso de 1 × KAPA Sybr® Fast Universal qPCR Mix (KAPA Biosystems, MA, EE.UU.), 400 nM de cada iniciador (Microsynth, Balgach, Suiza), DEPC-agua (Roth, Karlsruhe, Alemania) y 2 μl de plantilla de ADN. La PCR de amplicón de ciclo bajo se realizó utilizando un termociclador Labcycler (SensoQuest, Göttingen, Alemania) con el siguiente programa: desnaturalización a 95°C durante 5 min seguida de 10 ciclos de 95°C durante 30 s, recocido a 55°C durante 30 s y extensión a 72°C durante 30 s. Un último paso de extensión se realizó a 72°C durante 5 min. Los productos de la PCR de cada muestra con los cuatro pares de cebadores diferentes (fs0, fs1, fs2 y fs3) se mezclaron y posteriormente se purificaron con 0,8 × Agencourt AMPure XP Kit (Beckman Coulter, CA, EE.UU.) siguiendo las instrucciones del fabricante. Los productos de PCR purificados fueron resuspendidos en 30 μl de agua DEPC.
La PCR del índice Nextera XT se realizó en 50 μl de mezcla de PCR compuesta por 1 × KAPA Sybr® Fast Universal qPCR Mix (KAPA Biosystems), 5 μl de índices Nextera forward (N7XX) y reverse (S5XX) y 15 μl de producto de PCR previamente purificado. La PCR de índice se realizó como se ha descrito anteriormente, pero con 8 ciclos en lugar de 10. Los productos de PCR se purificaron como se ha descrito anteriormente con 1 × Agencourt AMPure XP Kit (Beckman Coulter).
La pureza de los productos de la PCR índice se comprobó con el Chip de Alta Sensibilidad de Bioanalyzer (Agilent Technologies, Santa Clara, EE.UU.). La concentración de los productos de PCR índice se determinó con el Fluorómetro Qubit® dsDNA HS Assay Kit y por qPCR con el KAPA Library Quantification Kit for MiSeq Illumina® (KAPA Biosystems) siguiendo las instrucciones del fabricante. Las muestras se agruparon añadiendo 0,75 nM de cada muestra a la biblioteca, que posteriormente se concentró a 4 nM con el Kit 1 × Agencourt AMPure XP (Beckman Coulter).
Secuenciación de amplificadores de alto rendimiento
La secuenciación de alto rendimiento de los amplicones de PCR del gen del ARNr 16S con extremo en paréntesis (2 × 300 nt) se llevó a cabo en una única ejecución multiplexada en un MiSeq de Illumina (Illumina® Inc., San Diego, EE.UU.; software v2.4.1.3) en la GDC. Se obtuvieron un total de 4.095.381 secuencias a partir de 23 muestras. Las secuencias de las réplicas de campo A-C se fusionaron dando como resultado 8 muestras: J0: 462.608 lecturas; J1: 543.977; J2: 107.085; J3: 88.383; J4: 961.545; J5: 198.260; J6: 1.108.814; J7: 624.709. Todos los resultados basados en el HTS reportados en este estudio se basan en estas 8 muestras. El procesamiento de los datos del gen del ARNr 16S se realizó siguiendo el proceso diseñado en la CD alemana (Tabla suplementaria 2). Las secuencias se agruparon a un nivel de similitud de secuencia del 97% y se definieron como Unidad taxonómica operativa (UTF) con Uparse en Uchime (Edgar, 2013). Sólo las OTUs detectadas más de 3 veces fueron consideradas para el análisis de la comunidad, reduciendo la complejidad de 637 a 539 OTUs. Las lecturas de secuencia obtenidas en este estudio fueron depositadas en el Archivo Europeo de Nucleótidos (ENA) con el número de proyecto PRJEB9478 (www.ebi.ac.uk/ena/data/view/PRJEB9478).
PCR cuantitativa en tiempo real
La cuantificación de las copias del gen del ARNr 16S se realizó mediante una PCR cuantitativa en tiempo real (qPCR) en un sistema ABI 7500 (Applied Biosystems) en la GDC. 20 μl de la mezcla de reacción qPCR contenía 1 × KAPA Sybr® Fast Universal qPCR Mix (KAPA Biosystems), 400 nM de cada cebador, hacia adelante B341F_fs0 y hacia atrás B805R_fs0 (Microsynth, Balgach, Suiza), DEPC-agua (Roth, Karlsruhe, Alemania) y 1 μl (0,2-43 ng/μl) de plantilla de ADN por reacción. Se incluyeron triplicados de controles sin plantilla, que contenían agua tratada con DEPC. Se realizaron ciclos térmicos utilizando el siguiente programa: desnaturalización a 95°C durante 10 min, seguido de 40 ciclos de 95°C durante 20 s, recocido a 55°C durante 30 s, y extensión y adquisición a 72°C durante 1 min. El último paso de extensión se realizó a 72°C durante 5 min.
Como norma bacteriana, se utilizaron series de dilución de productos de PCR derivados del ADN extraído de la cepa de control Methylococcus capsulatus (cepa Bath; cortesía del Prof. Svenning, Universidad de Tromsø, Noruega) en un rango entre 2,12 e1 y 2,12 e8. El número de copias del gen 16S rRNA se calculó a partir de las curvas estándar, asumiendo que la masa molecular media de una molécula de ADN de doble cadena es de 613 g mol-1.
Tema original:
Related posts
Warning: Undefined variable $sizeV in /home2/o8g4r1s7/public_html/homclinic.com/wp/wp-content/themes/betheme/functions/theme-functions.php on line 1284

Your quality seal

Protocolo para La Viruela Símica